
Enfermedad de Parkinson
Generalidades
La EP es un trastorno neurodegenerativo complejo con aparición en edad adulta, además de ser la segunda patología neurodegenerativa más común luego de Alzheimer. Es de etiología desconocida pero puede explicarse como una combinación de factores genéticos y ambientales. Su base anatomopatológica es una pérdida progresiva de neuronas dopaminérgicas de la sustancia negra pars compacta, SNpc, del mesencéfalo, e inclusión de cuerpos de Lewy formados por proteína alfa-sinucleina insoliblres anormalmente plegadas. Lo que genera es denervación de las proyecciones de la SNpc al núcleo estriado provocando alteración fisiológica de ganglios basales. En la clínica se encuentra la tríada motora: acinesia, temblor en reposo y rigidez, por lo que se considera un trastorno motor. Aunque recientemente se han descubierto manifestaciones no motoras como apatía, deterioro cognitivo o síntomas disautonómicos, que generan un alto impacto en la calidad de vida de los pacientes. El diagnóstico de sospecha es clínico y no existen marcadores biológicos específicos. Hay pruebas complementarias como la resonancia magnética craneal, RMc, o DaTSCAN. No existe tratamiento curativo pero si terapéutico y de control sintomático en cada fase.

Historia
En el año 2500 a.C. se describían diversos tipos de tremores y parálisis en textos de medicina Ayurvérdica, que probablemente hicieron referencia a EP. Siglos después, Galeno refiere en sus compendios, temblores y alteraciones de la marcha que cobraron importancia hasta la edad media. La EP conocida como hoy en día nace en 1817 con el cirujano británico James Parkinson, cuando publica la monografía ''An essay on the shacking palsy''. Buscó integrar en un sólo trastorno las manifestaciones que en ese momento se consideraban entidades distintas. Luego, el neurólogo francés Charcot atribuye el epónimo Maladie de Parkinson, enriquece descripción clínica.A inicios de siglo XXm se dilucidan parte de los mecanismos patogénicos como descripción, en 1913, por el patólogo Friederich Lewy de inclusiones citoplasmáticas llamadas cuerpos de Lewy, que poseen alfa-sinucleína, seis años luego la degeneración de la SNpc por el neuropatólogo Konstantin Tretiakoff. Décadas después, Arvid Carlsson demuestra que el déficit de dopamina es un trastorno neuroquímico subyacente a la enfermedad, que sirve para ensayos con levodopa por Birmarkmayer y Hornykievicz. Luego, Cotzias convertirá al precursor oral de dopamina en el tratamiento eje de EP actualmente. La levodopa generó un abandono de técnicas ablativas quirúrgicas pero aparecieron luego complicaciones derivadas del uso crónico causando revitalización de técnicas neuroquirúrgicas, como palidotomoías, neurocirugía funcional. El ultrasonido focal de alta intensidad permite el renacimiento de abordaje lesional por medios no quirúrgicos.
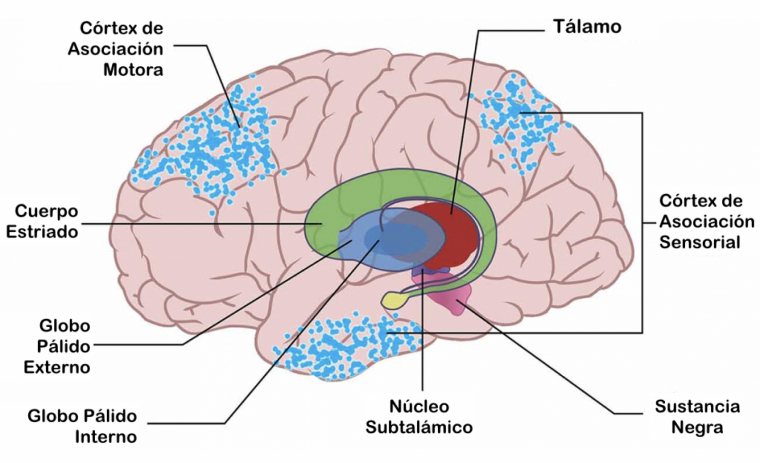
Fisiología de Ganglios Basales
La disfunción del sistema de ganglios basales, GB, es una base fisiológica de la EP, debido a la depleción de dopamina. Los GB son núcleos subcorticales que incluyen en el núcleo estriado, núcleo subtalámico y globo pálido externo e interno con sus conexiones con la SNpc, la sustancia negra pars reticulata SNr y el núcleo ventro-lateral del tálamo. Los núcleos se interconectan anatómica y funcionalmente, por proyecciones al tálamo y tronco cerebral conforman una red córtico-subcortical. Los GB se segregan en tres circuitos; límbico, motor y asociativo o cognitivo. La región dorso-lateral forma el circuito motor proyectándose sobre el córtex motor primario y área motora suplementaria, porción ventromedial de los núcleos general el circuito límbico y conecta con áreas límbicas corticales, la porción localizada entre ambas proyecta en córtex prefrontal creando el circuito asociativo.Alteraciones de ganglios basales características de EPLa depleción dopaminérgica resulta en alteraciones neurofisiológicas de la actividad de los GB que subyacen a los síntomas cardinales de la enfermedad. Por la falta de dopamina se produce potenciación de la vía indirecta inhibidora sobre la directa facilitadora, que se traduce en movimientos constantes por incremento de tasa de descarga de NST y complejo Gpi/SNr e inhibición tálamo-cortical. El aumento se debe a la depleción dopaminérgica, mientras que la discinesia por levodopa se asocia a menor actividad en Gpi y NST. Asimismo, la falta de dopamina genera que neuronas en GB descarguen de manera oscilatoria en lugar de una activación tónica fisiológica.

Epidemiología y Etiología
Prevalencia: 0.3% de la población general y el 1% es en mayores de 60 años. Incidencia: 8-18 por 100.000 habitantes/año. Proporción: es más frecuente en hombres que en mujeres por efecto protector de estrógenos. Etiología: el principal factor es el envejecimiento, también puede ser por mutación genética específica. En EP de inicio joven, menor a 40 años (5% del total de EP), tienen mayor probabilidad que sea por origen genético y se asocia a herencia autosómica recesiva. En personas con EP de inicio anterior a los 45 años, la mutación más común es la del gen de la parkina, presente en el 50% de los casos familiares y en el 15% de los esporádicos.Pronóstico: la forma rígido-acinética serían factores predictores de una progresión más rápida, mientras que la forma de inicio tremórico tiene mejor pronóstico
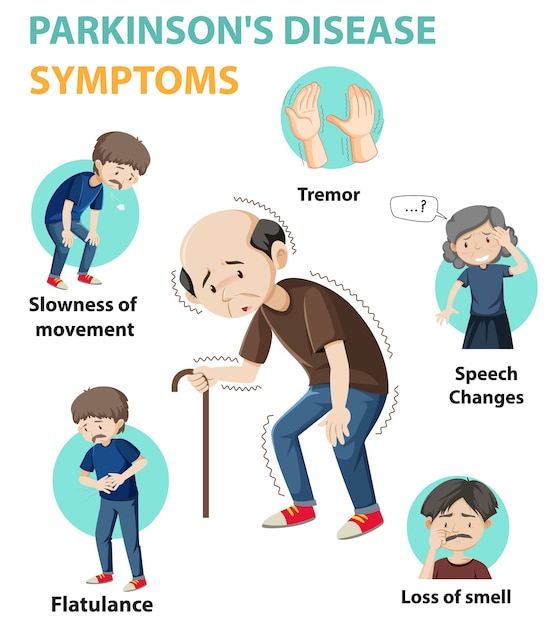
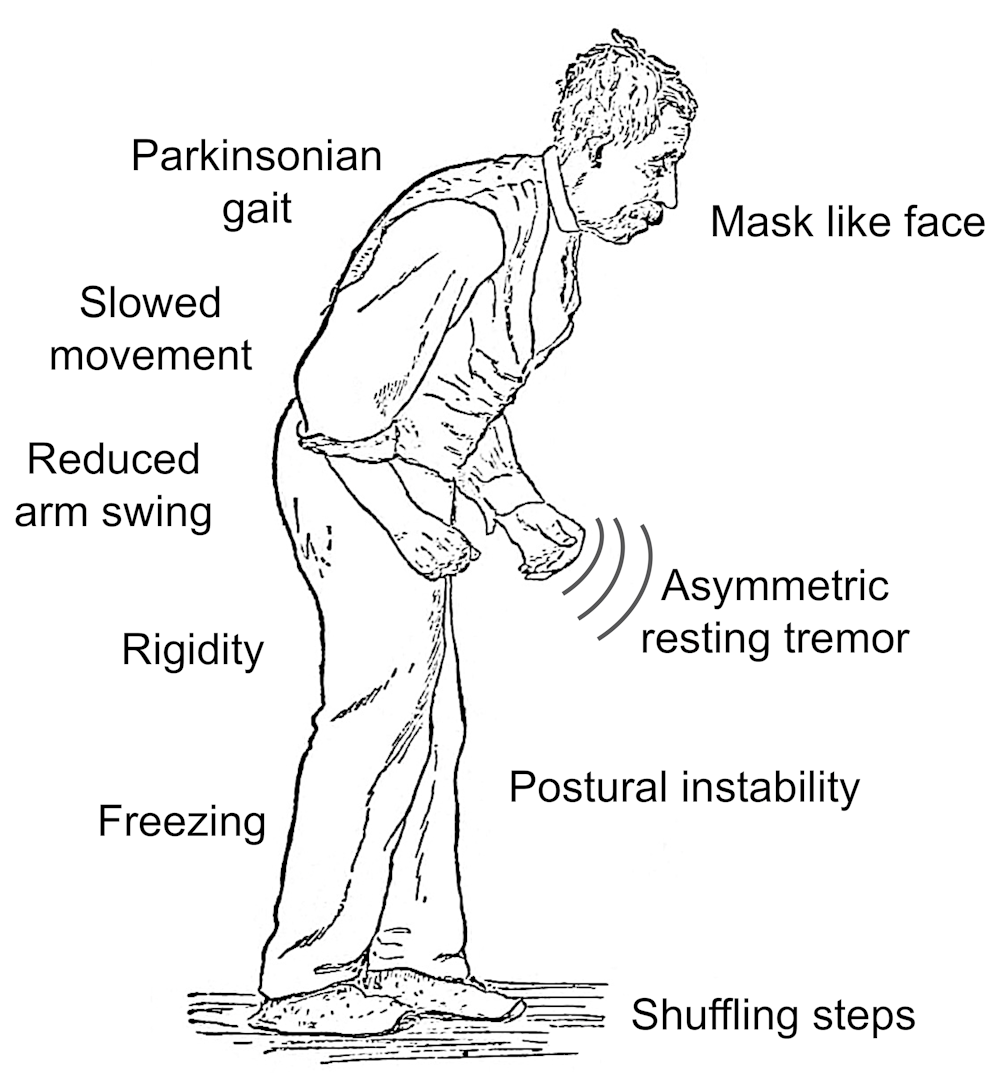
Clínica
Manifestaciones motoras
Tremor de reposo (70% de los casos) grosero y de gran amplitud, frecuencia de 4-6HzRigidez en rueda dentada Acinesia Fatigabilidad Decremento progresivo de amplitud en movimientos repetitivos
Manifestaciones no motoras
Apatía DepresiónAlteraciones del sueñoDisfunción autonómica o síntomas sensitivos HiposmiaConstipaciónHipersomnia diurna Puede existir alteración en la visualización de coloresPeríodo prodrómico: hipersomnia diurna, alteración en visión de color, fatiga, apatía, dolor central.Estos síntomas se han relacionado con la distribución de los cuerpos de Lewy en el sistema nervioso, específicamente en el sistema nervioso periférico, bulbo olfatorio, tronco cerebral, sistema límbico y corteza cerebral, según el mapa de sinucleopatía descrito por Braak y Braak.
Progresión
Las manifestaciones motoras se generalizan conforme avanza la enfermedad, aunque mantienen algo de asimetría. A mayor denervación dopaminérgica hay menor capacidad de almacenar, recaptar y liberar la levodopa que se ingiere por lo que la concentración de dopamina cerebral es errática generando complicaciones como:Deterioro fin de dosis cuando la toma no mantiene el efecto hasta la siguiente toma Fallo del efecto de una dosis Bloqueos repentinos AnsiedadSudoración profusa Bradipsiquia Fatiga Acatisia Disquinesias: distonía , disquinesias de pico de dosis cuando el nivel de dopamina es máximo Disfasias cuando el paciente está pasando del estado de bloqueo a estado ON, o viceversa, y varía la concentración de dopamina.Conforme progresa pueden haber manifestaciones axiales Alteración reflejos posturales Imantación de la marchaHipofoníaDisartriaDisfagiaTras 20 años de enfermedad: el 83% presenta demencia. Con deterioro cognitivo, atención, función ejecutiva, memoria, función visuoespacial, cambios afectivos, alucinaciones de predominio visual, apatía. Todo esto conlleva a una pérdida de autonomía.

Tratamiento
EP precoz
Aspectos motoresFármaco de elección: levodopaRasagilina (IMAO-B) Agonistas dopaminérgicos no ergóticos, por ejemplo la rotigotina, pramipexol o ropinirolPuede iniciarse con rasagilina en monoterapia, pero puede combinarse con agonistas dopaminérgicos o levodopa. Aspectos no motoresPrevio al uso de un medicamento específico deben realizarse optimización de fármacos dopaminérgicos por los síntomas causados debido al estado hipodopaminérgico. Si no hay mejoría debe realizarse tratamiento específico.
EP avanzada
El uso de fármacos de liberación prolongada, fragmentación de dosis orales de levodopa o añadir terapias potenciadoras de la misma pueden aminorar las fluctuaciones en el paciente. La safinamida aumenta el tiempo ''on'' sin incremento de discinesias. En pacientes que no son óptimamente controlables con terapia médica convencional se puede utilizar la estimulación cerebral profunda, la bomba de apomorfina, bomba de levodopa/carbidopa enteral y ultrasonido focal de alta intensidad.

Diagnóstico
Es de sospecha clínica, el diagnóstico definitivo se realiza con confirmación de hallazgos neuropatológicos como pérdida neuronal en SNc, cuerpos y neuritas de Lewy presentes, que puede realizarse únicamente una vez ha fallecido el paciente. El Banco de Cerebros del Reino Unido provee criterios clínicos para el diagnóstico: Presencia de parkinsonismo (bradicinesia, u otro signo motor)Exclusión de otras causas justificantes, descartadas por los antecedentes del paciente y examen físico neurológico Existencia de datos característicos de la EP que apoyen el diagnóstico. En la anamnesis debe incluirse el curso lentamente progresivo de la clínica cardinal motora, y su distribución asimétrica, interrogar de medicación actual (puede inducirse por neurolépticos, para flatulencias, antivertiginosos, entre otros). La exploración neurológica debe descartar signos atípicos sugestivos de otras causas de parkinsonismos como parálisis supranuclear de la mirada, alteraciones cerebelosas o del equilibrio, déficits cognitivos clínicamente significativos de inicio precoz, signos corticales, disfunción autonómica.