
Cystic Fibrosis
What is Cystic Fibrosis
Cystic Fibrosis or better known as CF, is an inherited genetic disorder that causes progressive and severe damage to the digestive system, lungs, and many other organs in the body. The disorder is defined by a buildup of abnormal mucus that can damage many of the body's organs. Mucus is a secreted substance that protects and lubricates the linings of airways, reproductive system, digestive system and other tissues and organs.

Normally, Mucus is thin and slippery. However, in terms of people with CF, a defective gene prompts the cells to produce sticky and thick Mucus. That creates a significant blockage in the airways, inducing complications with breathing and infections in the lungs. These bacterial infections generate chronic wheezing, coughing, and swelling. Moreover, as time goes on, the accumulation of mucus and infections result in the formation of scar tissue (fibrosis), perpetual lung damage, and cysts in the lungs. The following link has a detailed picture of how the airways, and other organs get clogged/affected with mucus.
Cause of Cystic Fibrosis
The cause of Cystic Fibrosis is a mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. The CFTR gene codes a protein called the CFTR protein.
The CFTR protein is a particular type of protein called an ion channel. Ion channels move molecules or atoms that have a positive or negative charge from outside the cell to inside, or inside the cell to outside. The CFTR protein moves a negatively charged chlorine inside and out of the cells. To get out of the cell, the chlorine ions go through the center of the tube created by the CFTR protein. Once out of the cell, the chlorine ions attract a layer of water. That is important because the water layer allows cilia (tiny hairs) on the lungs to sweep back and forth, which makes the mucus move up and away from the airways.
However, in people with Cystic Fibrosis, the mutation in the CFTR gene disrupts the function of the chlorine channels, preventing them from managing the flow of the chlorine ions and water across the cell membrane. That causes the chlorine ions to be trapped inside the cell, and the water no longer attracts itself to the outside of the cell. Due to this, there is less water outside the cell than normal, causing mucus to become condensed and dehydrated. As a result, any cells that line in the passageways of the lungs, pancreas, and other organs produce this type of mucus. This particular mucus flattens the cilia in the lungs and restricts it from properly sweeping due to the weight of mucus. Therefore the airways get clogged and various ducts, causing the characteristic signs and symptoms of CF.
As a result, any cells that line in the passageways of the lungs, pancreas, and other organs produce this type of mucus. This particular mucus flattens the cilia in the lungs and restricts it from properly sweeping due to the weight of mucus. Therefore the airways get clogged and various ducts, causing the characteristic signs and symptoms of CF.
As years past, scientists have used multiple different ways of grouping these different mutations into different classes. The most recent CFTR classification system groups mutations by the problems that they cause in the production of the CFTR protein:
Protein production mutations (Class 1)
Protein processing mutations (Class 2)
Gating mutations (Class 3)
Conduction mutations (Class 4)
To learn more about the CFTR, click the links below.
1
Inheritance Pattern (How a person gets CF)
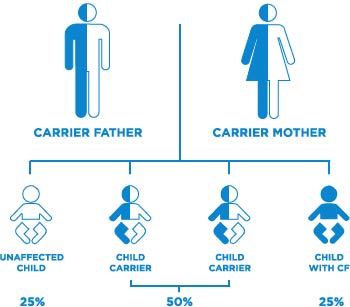
As stated in the beginning, Cystic Fibrosis is an inherited disorder. Its type is called an autosomal recessive disorder. That means that the mutated gene is not solely inherited from either mother or father, but the mutated gene is from both parents. When someone has one copy of the mutated gene, the functional (dominant) gene can compensate for the defect (recessive) gene, and the person will not have CF. However, just because they are healthy, that does not mean they cannot be carriers.
If two parents are carriers of the defect gene, their child has a:
25% (1 in 4) chance that they can have CF
50% (1 in 2) chance that there child will be carriers, but not have CF
25% (1 in 4) chance that the child is not a carrier, and will not have CF
If a person with CF has a child with a carrier of CF, their child has a:
50% (1/2) chance their child will have CF
50% (1 in 2) chance that there child will be carriers, but not have CF
If both parents have CF, their child has a:
100% chance of getting CF
Other Inheritance Patterns that are not genetic but more noticeable is that CF is more dominant in European Blood Lines than any other race. Most CF cases across the globe are from European descent, and reside mostly in North America.
For more information on the exact numbers and precentages for people affected by CF, please press the link.
Signs and Symptoms
Some signs and symptoms of CF that is common amongst many is:
1. Clubbing of fingers and toes caused by less oxygen getting to the hands and feet
2. Fever, which may include night sweats
3. Gastrointestinal traits, such as prolonged diarrhea, severe gastric pain, prolonged diarrhea, or constipation
4. Jaundice, or yellow skin, for an abnormally long period of time after birth
5. Low body mass index (BMI) or being underweight
6. Muscle and joint pain
7. Delayed growth or puberty
8. Abnormally Salty skin/sweat
9. Bulky, greasy, bowel movements
10. Sinus and/or lung infections or pneumonia
How is CF Diognosed
In many provinces in Canada, and around the world, CF can be diagnosed in multiple ways. early diagnosis is done through newborn screening tests.
IRT: Early diagnosis is done through an Immunoreactive trypsinogen (IRT) test in newborn screening. The screening checks for abnormal levels of IRT protein in a newborn, and High-Levels of IRT can be a sign that the baby has CFR. However, this test does not guarantee a 100% confirmation, so further test are ran to confirm it.
Sweat Test: A Sweat Test is the most commonly used test for CF. Since people in CF have a very salty sweat, the test checks to see an increased levels of sodium in it. It is preformed by using a chemical that makes the skin sweat when stimulated by a weak electric current. Sweat is gathered on a pad/paper and then analyzed. A diagnosis of cystic fibrosis is made if the sweat is saltier than normal.
CT Scans: A CT scan creates precise images of the body by using a combination of X-rays taken from many disparate directions. These images allow the doctor to view your internal organs, such as the liver and pancreas, making it simpler to assess the extent of organ impairment caused by CF.
Treatments
Though there is no cure for cystic fibrosis, treatments can relieve symptoms, decrease the complexities of CF, and improve quality of life. Monitoring, fast, and aggressive intervention is recommended to slow the progression of CF, which can raise the life expectancy of a patient. Overseeing cystic fibrosis is complex, so getting treatment at a center with a team of doctors and medical specialists trained in CF can help treat your condition.
Various Types Medicine For CF Treatments
Mucus-thinning prescriptions can help you or your child cough out the mucus, which can improve lung function. This can also be a This website link attached will help direct you into properly administrate this medication, and others safely.
Common Mucus Thinners for CF:
Dornase Alfa: acts like scissors by splitting up the long DNA strands contained in white blood cells. By cutting the strands into shorter pieces, Dornase Alfa helps break up dense and sticky mucus.
Hypertonic Saline: Sterile saline that increases the level of sodium in the airways to attracts water to thin the mucus
Nebulizers also are a way of treatment, to help remove mucus out of airways. For more info, please click the link.
Antibiotics help to treat and reduce lung infections. The website attached can help you discern which group which type of antibiotics are available to use for CF, and which ones are most likely to be prescribed to you.
Anti-inflammatory medications to decrease swelling in the airways.
Inhaled medications called bronchodilators help keep your airways open by loosening the muscles around your bronchial tubes. This list includes some of the various types of bronchodilators that can be prescribed to you or your child.
Albuterol (branded names include Ventolin and Proventalin)
To learn more about how to use bronchodilators or to get insight on the side effects and more, click the links.
Xopenex (levalbuterol)
Combivent
Acid-reducing medications to help pancreatic enzymes work better
Oral pancreatic enzymes to help your digestive system absorb nutrients
Stool softeners to prevent constipation/bowel difficulties
Acid-reducing prescription to help pancreatic enzymes work more efficiently
Explicit drugs for diabetes or liver disease, when suitable
Exercise
Physical activities have proven to help enhance strength and overall internal health in people with Cystic Fibrosis. It has also proven to help clear the mucus from the lungs, which will help relieve symptoms and pains people go through. To learn how to exercise while being cautious, click the following link to have articles and blogs to help you to achieve this!
Airway Clearance
Airway clearance techniques or (CPT) Chest Physical Therapy can aid mucus obstruction and help to decrease infection and inflammation in the airways. These techniques extricate thick mucus in the lungs, making it easier to cough out.
High-Frequency Chest Wall Oscillation: The High-Frequency Chest Wall Oscillation, or the Vest is an inflatable vest that is connected to a machine that causes the vest to vibrate. The vibrations help loosen the mucus in the airways. A video attached to this block will teach and demonstrate how preform this on your child
(CPT) Chest Physical Therapy Link
Surgery
Some surgeries can be used to help relieve CF, and or save a life if the condition progresses.
Nasal/sinus surgery: Doctor can recommend surgery to remove nasal polyps that hinder breathing. Sinus surgery can be done to treat intermittent or chronic sinusitis.
Feeding Tube: Cystic fibrosis intervenes with digestion, so you can't consume nutrients from food accurately. A doctor may suggest using a feeding tube to deliver more nutrition. This tube can be a temporary tube inserted into your nose and to your stomach, or the tube may be surgically implanted in the abdomen. The tube can be used to give extra calories throughout the day or night and does not restrict eating by mouth.
Lung Transplant: If you have critical breathing problems, life-threatening lung difficulties or increasing resistance to antibiotics for lung infections, lung transplantation could be an option. Because bacteria panel the airways in diseases that cause permanent widening of the large airways (bronchiectasis), both lungs need to be replaced.
*Cystic fibrosis does not recur in the newly transplanted lungs. However, other difficulties linked with CF, such as sinus infections, diabetes, pancreas conditions could still happen after the transplant.*
History of Disorder
The closest earliest mention of cystic fibrosis medical texts was around 1595. These texts linked salty skin and damage to the pancreas (symptoms in CF) with death in infants and youthful children.
In 1938, an American pathologist, Dr. Dorothy Andersen, presented the first description of the CF in the medical literature, calling it “cystic fibrosis of the pancreas” based on her autopsy conclusions of children who died of malnutrition. Other physicians of that era referred to the disease as “mucoviscidosis,” due to the appearance of thick mucus.
The CFTR protein was not noticed as the responsible gene until the 1980's, However , every since its discovery the years ahead, (after discovery and now) has new therapies have been introduced, leading to a climactic increase in survival.
The websites below shows more information on the survial rate
Current Research/Discoveries on Cystic Fibrosis
A study found by mayoclinic.org shows in the treatment page of their website (for CF), new Medications that target gene mutations, including a new medication that combines three drugs to treat the most common genetic mutation causing CF and is considered a major achievement in treatment
For those with cystic fibrosis who have certain gene mutations, doctors may recommend cystic fibrosis transmembrane conductance regulator (CFTR) modulators. These newer medications help improve the function of the faulty CFTR protein. They may improve lung function and weight, and reduce the amount of salt in sweat.
The FDA has approved these medications for treating CF in people with one or more mutations in the CFTR gene:
The newest combination medication containing elexacaftor, ivacaftor and tezacaftor (Trikafta) is approved for people age 12 years and older and considered a breakthrough by many experts.
The combination medication containing tezacaftor and ivacaftor (Symdeko) is approved for people age 6 years and older.
The combination medication containing lumacaftor and ivacaftor (Orkambi) is approved for people who are age 2 years and older.
Ivacaftor (Kalydeco) has been approved for people who are 6 months and older (Staff, Cystic fibrosis 2020).
Since so many discoveries happen worldwide on Cystic Fibrosis, it is very difficult to pin point which ones are important to put down. However, I put one of my favorite discoveires in a box for you to review. In that being said, I have put down ones website that inform people about research that goes on around the world.
Community support agencies
Some organizations in Canada are dedicated in educating people on people who live, struggle, and thrive inspire of complication from Cystic Fibrosis. Some of the most informative support communites in Canada Include:
Canada Helps (CF extention)